How to repair a DNA double strand break?
DNA double-stranded breaks (DSB) are recognized as the major genotoxic lesion of ionizing radiation. The use of ionizing radiation in medical diagnostics and therapy mandates an exact understanding of the biological processes surrounding the formation of radiation damage, its recognition, its repair, and of other cellular responses (e.g., apoptosis) to DSBs. Moreover, anti-cancer drugs, including a class of microbial secondary metabolites (e.g., bleomycin), topoisomerase inhibitors (Top1: camptothecin; Top2: etoposide) as well as inhibitors of poly (ADP-ribose) polymerase (PARP) also cause DSBs directly or indirectly. To potentially enhance the efficacy of treatment and to minimize side-effects it is crucial to understand the full range of cellular responses to this type of DNA damage. Thus, characterizing the cellular responses to DNA DSBs constitutes a major goal in biomedical research.
DSBs are also intermediates of normal cellular processes like DNA replication, meiosis, and immunoglobulin rearrangement. A single unrepaired DSB can be lethal for a cell or lead to disastrous consequences upon losing the affected chromosome. Misrepair of DSBs can result in chromosomal aberrations and other types of mutations. DNA repair is a major mechanism to generate genomic stability. Genomic instability is a hallmark for all forms of cancer. Hereditary cancer predisposition and DNA metabolism are obviously linked as indicated by numerous examples including Nijmegen Breakage Syndrome, ataxia telangiectasia, xeroderma pigmentosum, Bloom’s syndrome, and Lynch syndrome. Thus, DNA repair is not only a prerequisite for the evolution of cellular life, but it is also essential for ontogenic development.
Pathway usage during DSB repair and stalled replication forks
Several pathways ensure the repair of DSBs in eukaryotic cells. Recombination is the only inherently high-fidelity pathway to accomplish this, and it involves a complex series of events which are now beginning to be understood in eukaryotes (see Figs. 1 and 2). Other pathways like Single-Strand Annealing (SSA), Non-Homologous End-Joining (NHEJ), Pol Theta-Mediated End-Joining (TMEJ) and Break-Induced Replication (BIR) are error-prone, leading to mutations and loss of heterozygosity (Fig. 1). These DSB repair pathways are conserved in all eukaryotes with the exception of TMEJ, which is found only in a subset of eukaryotes (e.g., not in yeast but in mammals). In fact, recombinational repair is conserved in all forms of life known to date.

It is unclear how the usage of the four major DSB repair pathways are regulated. In collaboration with Dr. Markus Löbrich’s laboratory at Technical University Darmstadt (Germany), we identified an unexpected novel role of BRCA2 and RAD52 in inhibiting TMEJ in S and G2 cells, delaying TMEJ-mediated DSB repair until M-phase (Llorens-Agost et al. 2021 Nature Cell Biol). The critical insight that TMEJ happens in M-phase provides a compelling explanation for the observed synthetic lethality between defects in BRCA2 and RAD52, which is caused by the premature action of TMEJ in the S- and G2-phases leading to genomic rearrangements and mitotic catastrophe.
To elucidate the hierarchy of pathway usage at stalled replication forks, we isolated separation-of-function mutants in budding yeast Rad51 that effectively separate its function in HR from its function in fork protection and pathway choice. Two single amino acid changes in Rad51 have no effect on HR in spontaneous and DSB-induced recombination assays, but show a strong mutator phenotype and hyper-reliance on translesion synthesis, and fork regression upon stalling replications forks with methyl methanesulfonate (Meyer et al. 2023, in preparation). These results suggest that HR-mediated gap repair (Fig. 1) is a primary response after fork stalling and repriming.
Mechanism of homologous recombination in eukaryotes
Progress in identifying the components of recombinational repair (Fig. 2) has provided convincing evidence that this entire pathway is conserved (Kowalczykowski, Hunter, Heyer 2016 Cold Spring Harbor Press). Work with transgenic mice has also provided conclusive evidence that recombinational repair is active and important in mammalian cells. Interestingly, this pathway is also crucial for the repair of DNA crosslinking agents, another class of anti-cancer drugs, and in replication fork support (Fig. 1). Purification of the individual components of this DSB repair pathway and definition of the biochemical function of these proteins, alone and in conjunction, is crucial to understand the mechanism of recombinational repair. Based on the premise of evolutionary conservation of the DNA repair pathways, it appears that the basic mechanism of DSB repair will be highly similar in all eukaryotes. The situation in higher eukaryotes like mammals will undoubtedly be more sophisticated in details. Thus, we have decided to concentrate the mechanistic studies on the yeast S. cerevisiae, a model system for which many components are identified and genetic analysis is easily possible, and followed by validation of our insights in human cells.
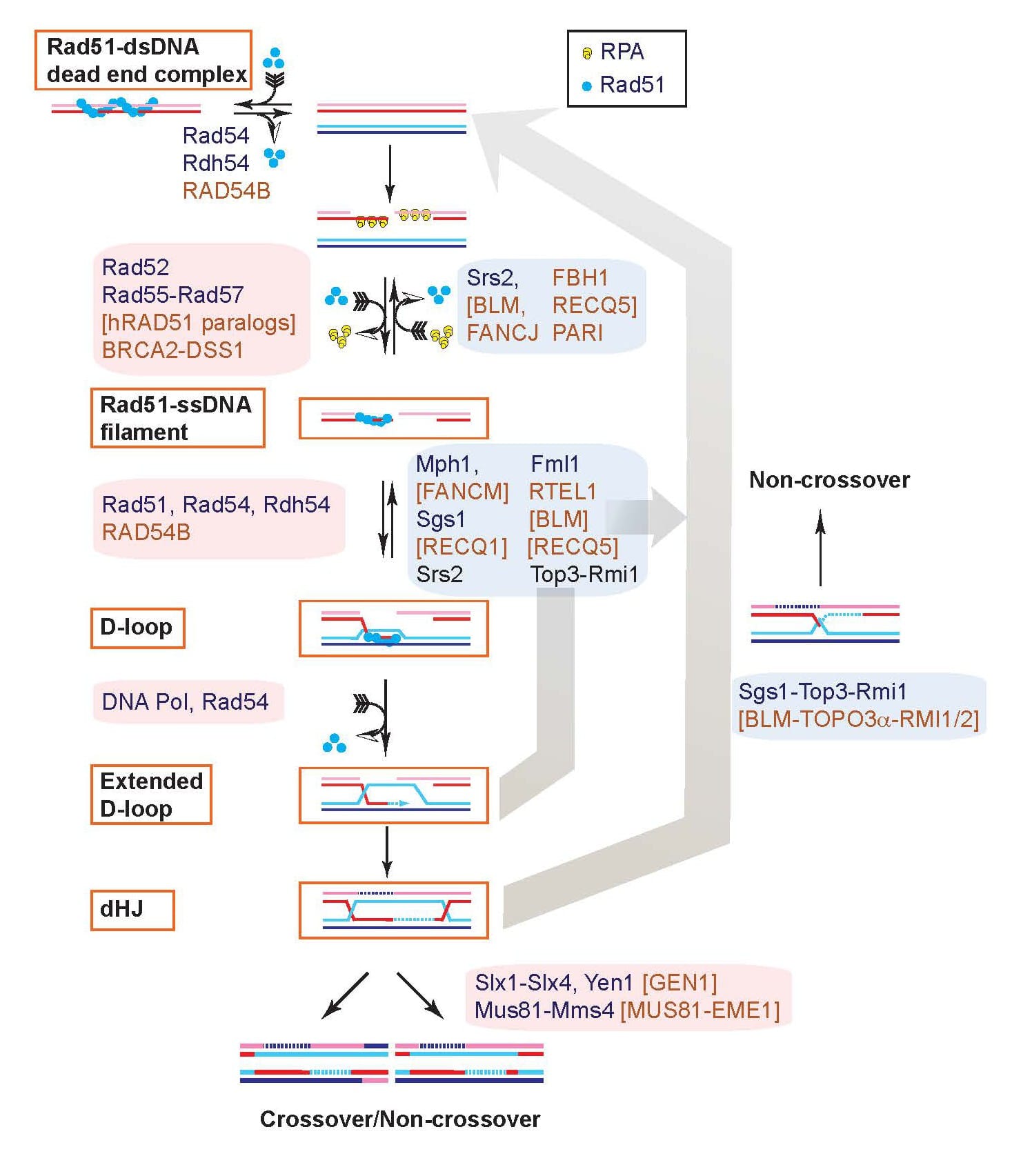
Combining biochemistry with newly developed proximity ligation assays to directly measure D-loop intermediates and D-loop extension we have delineated two pathways of D-loop reversal in vivo and showed that the majority of D-loops between perfectly matched sequences (save for a heterologous terminal flap) are reversed (Piazza et al. 2019 Mol Cell; Piazza et al. 2018 Meth Enzymol; Reitz et al., 2022 JOVE). These results provide also the biological significance for our earlier biochemical studies showing that Top3-Rmi1 and Srs2 can disrupt D-loops (Fasching et al. 2015 Mol Cell; Liu et al. 2017 Elife). In addition, these proximity ligation assays suggested a role for Rdh54 in regulating D-loop length. To address this question, we developed another innovative assay, the D-loop mapping assay, which allows for the quantification of D-loop length using bisulfite modification of the displaced DNA strand in D-loops and SMRT-PacBio DNA sequencing of individual DNA molecules (Shah et al. 2020a Elife). This technique enabled us to define a role of the Rdh54 (human RAD54B) motor protein in limiting D-loop size by counteracting the Rad54 motor protein (Shah et al. 2020b Elife). We are currently adapting the D-loop mapping assay to use in cells to determine the length of D-loops in cells and their dynamics (D-loop growth or migration). In collaboration with Dr. Markus Löbrich’s laboratory at Technical University Darmstadt (Germany), we are currently extending these studies to the human RAD54 and RAD54B proteins.
In biochemical experiments (Wright et al. 2014 Mol Cell), we discovered that long (>600 nt) Rad51-ssDNA filaments can invade multiple duplex target molecules simultaneously and later formally proved the existence of multi-invasion intermediates in vitro (Piazza et al. 2017 Cell). Using a designed reporter system in budding yeast, we demonstrated that multi-invasions also form in cells using additional novel proximity ligation assays (Piazza et al. 2017 Cell, Reitz et al. 2023 Genes Dev). Multi-invasions are actively suppressed by several independent pathways involving the structure-selective endonuclease Rad1-Rad10 (human XPF-ERCC1) as well as Sgs-Top3-Rmi1, Mph1, and Srs2 (Piazza et al. 2017 Cell). Multi-invasion intermediates are processed by structure-selective endonucleases including Mus81-Mms4, Slx1-Slx4, and Yen1 to yield genome rearrangements and additional DSBs that can lead to further genomic rearrangements involving repeated DNA (Piazza et al. 2017 Cell, Reitz et al. 2023 Genes Dev). Multi-invasion recombination can lead to cascades of rearrangements via two independent mechanisms that depends on the exact genomic sequence context. The predominant pathway, MIR1, does not require displacement DNA synthesis by polymerase delta, in contrast to the canonical HR pathway, and leads to the formation of secondary DSBs, explaining how the repair of a single DSB can lead to additional rearrangements.
The dsDNA motor protein RAD54
Our mechanistic work started with the Rad54 protein, one of the most crucial DSB repair proteins (Clever et al. 1997 EMBO J; Schmuckli-Maurer and Heyer 1999 Mol Gen Genet; Clever et al. 1999 Yeast; Schmuckli-Maurer and Heyer 2000 Chromosoma; Schmuckli-Maurer et al. 2003 NAR; Kiianistia et al. 2006 PNAS; Li et al. 2007 NAR; Li and Heyer 2009 NAR; Wright and Heyer 2014 Mol Cell).
To further characterize its role in recombination, we developed in vitro recombination reactions using purified human or yeas RPA, Rad51, and Rad54 proteins (Mazin et al. 2000 Mol Cell; Solinger et al. 2001 JMB; Solinger and Heyer 2001 PNAS), either alone or in combination with the proteins required for HR-associated DNA synthesis (PCNA, RFC, Pol delta) (Li et al. 2009 Mol Cell; Sneeden et al. 2013 NAR). The Rad54 protein interacts specifically with Rad51 in the presynaptic filament to stimulate homologous pairing and DNA strand exchange in the synaptic (Mazin et al. 2000 Mol Cell; Solinger et al. 2001 JMB) and post-synaptic phases (Solinger and Heyer 2001 PNAS) of recombination. Our initial efforts focused on the role of the dsDNA-dependent ATPase activity of Rad54 (Kiianitsa et al. 2002 JBC), and we were first to demonstrate that Rad54 can disassemble Rad51:dsDNA filaments (Solinger et al. 2002 Mol Cell). Later genetic and cytological evidence suggested a role for Rad54 after Rad51/Rad52/Rad55/57. We published a unifying model of Rad54 functions: Rad54 is a heteroduplex DNA pump that turns over Rad51 after DNA strand exchange and at the same time intertwines the invading and the complementary strands in the D-loop (Wright and Heyer 2014 Mol Cell) (Fig. 3). The model explains why Rad54 is required for access by DNA polymerases to the 3’-OH end of the invading strand in the D-loop (Li and Heyer 2009 NAR). Central predictions from this model have since been validated by electron-microscopic analysis (Tavares et al. 2019 Nat Commun).

The Mus81-Mms4 (MUS81-EME1) structure-selective endonuclease
Mus81/Mms4 is a DNA structure-specific endonuclease that we discovered in a two-hybrid screen using Rad54 as a bait (Interthal et al. 2000 Mol Gen Genet). Further genetic analysis provides strong arguments that Mus81/Mms4 functions late in recombination in a resolution pathway that is parallel to Sgs1/Top3/Rmi1 (Fabre et al. 2002 Mol Cell). These studies highlighted a novel type of synthetic lethality between gene defects in a single non-essential molecular pathway with reversible steps (Zinovyev et al. 2013 PLoS Comp Biol). Mus81-Mms4 is important for the recovery of stalled and/or broken replication forks (Heyer et al. 2003 TiBS), and our detailed kinetic analysis of the enzyme provided the biochemical basis for understanding its biological role in processing DNA junctions (Ehmsen and Heyer 2008 NAR, 2009 NAR; Schwartz et al. 2012 Mol Cell Biol; Mukherjee et al. 2014 NAR).
Rad51 paralogs
All eukaryotes have Rad51 paralogs that function in conjunction with Rad51 in HR. Their role remained elusive for long, and we identified the first specific function of the Rad51 paralogs in showing that Rad55-Rad57 stabilize the Rad51 filament against disruption by the anti-recombinogenic helicase Srs2 (Liu et al. 2011 Nature). Our current efforts are directed towards establishing a structure for the Rad51-Rad51 paralog co-filament and determining the functions of the putative ATPases of the Rad55 and Rad57 paralogs.
BRCA2
BRCA2 is a prominent tumor suppressor in breast, ovarian, prostate, and pancreatic cancer as well as additional malignancies. The BRCA2 protein is unusually large with 3,418 amino acids, and our laboratory was first together with the West and Kowalczykowski laboratories to achieve the purification of the full-length human BRCA2 proteins (Liu et al. 2010 Nat Struct Mol Biol). We showed directly that human BRCA2 loads RAD51 on RPA coated ssDNA and that its co-factor DSS1 stimulates this activity. We further defined the mechanism by which DSS1 stimulates BRCA2 and found that BRCA2 binding to DSS1 and ssDNA favors the transition of BRCA2 oligomers to monomers, its presumptive active form (Le et al. 2020 NAR). Currently, we are defining the self-interaction sites of BRCA2 and how self-interaction is regulated. In addition, we are elucidating the DNA binding properties of full-length BRCA2 and its individual DNA binding elements.
Regulation of DNA repair by DNA damage checkpoints
DNA damage checkpoints coordinate the cellular responses to DNA damage including transient cell cycle arrest and replication slow down, transcriptional induction of a large array of genes, and – in higher eukaryotes – programmed cell death. Defects in this signal transduction pathway lead to major radiosensitivity in all organisms studied and to hereditary cancer predisposition in humans, as exemplified by the syndrome ataxia telangiectasia (AT). A growing number of components of this pathway has been isolated, primarily through efforts in yeast model systems. However, the mechanism for how the checkpoints recognize DNA damage and elicit cellular responses is almost completely unknown. Seminal work with cells derived from AT patients suggested DNA repair defects in such cells that did not eliminate but somehow misguide DNA repair of DSBs.
We entered the checkpoint field based on the premise that the DNA damage sensing capability of checkpoints might provide a direct way for the DNA repair systems to be recruited to the sites of DNA damage. In particular, the biochemical properties of the DSB recombinational repair pathway suggested that DNA damage recognition is a major problem. Thus, we have directly analyzed the components of this pathway to determine whether they are substrates for the DNA damage checkpoints. We found that the Rad55 protein is specifically phosphorylated in response to a variety of DNA damages including ionizing radiation (even a single DSB) dependent on an active checkpoint by the Rad53 and Mec1 kinases (Bashkirov et al. 2000 MCB; Herzberg et al. 2006 MCB; Janke et al. 2016 NAR). Thus, we have established that both systems, checkpoint control and DNA repair, are directly linked.
Rad55 phosphorylation is a terminal checkpoint event and provides a unique opportunity to analyze the regulation of the upstream signal transduction cascade. In trying to understand how the checkpoint kinases are regulated, we demonstrated that Dun1 kinase is directly phosphorylated by Rad53 kinase after genotoxic stress (Bashkirov et al. 2003 MCB). The specificity of this interaction is determined by the FHA domain of Dun1. The interaction between activated Rad53 kinase and Dun1 is highly transient and destabilized by autophosphorylation of Dun1 after Dun1 is activated by trans-phosphorylation by Rad53. This provides a mechanism for signal amplification, as one activated Rad53 kinase molecule can potentially activate many Dun1 kinase molecules. These studies also revealed a Rad53-independent role of Dun1 kinase. Moreover, we identified the existence of a truncated DNA damage response that activates Mec1 kinase using Rad55 as an output signal but constrains the signal locally without activating Rad53 kinase (Janke et al. 2010 NAR).